1. Introduction
BindWeb is an easy-to-use online server for ligand binding residues and pockets prediction from protein structures, it is designed for ligand-specific and ligand-general binding residues and pockets prediction. It consists of two deep learning-based methods: a graph neural network(GNN) based method GraphBind1 and a hybrid convolutional neural network (CNN) and bidirectional long short-term memory network (biLSTM) based method DELIA2, which are integrated to make more accurate ligand binding pockets prediction. In addition, we add a module for clustering predicted binding residues into binding pockets, which is useful for more than one pocket predicted in the same query structure. The pipeline of BindWeb is shown in Figure 1.
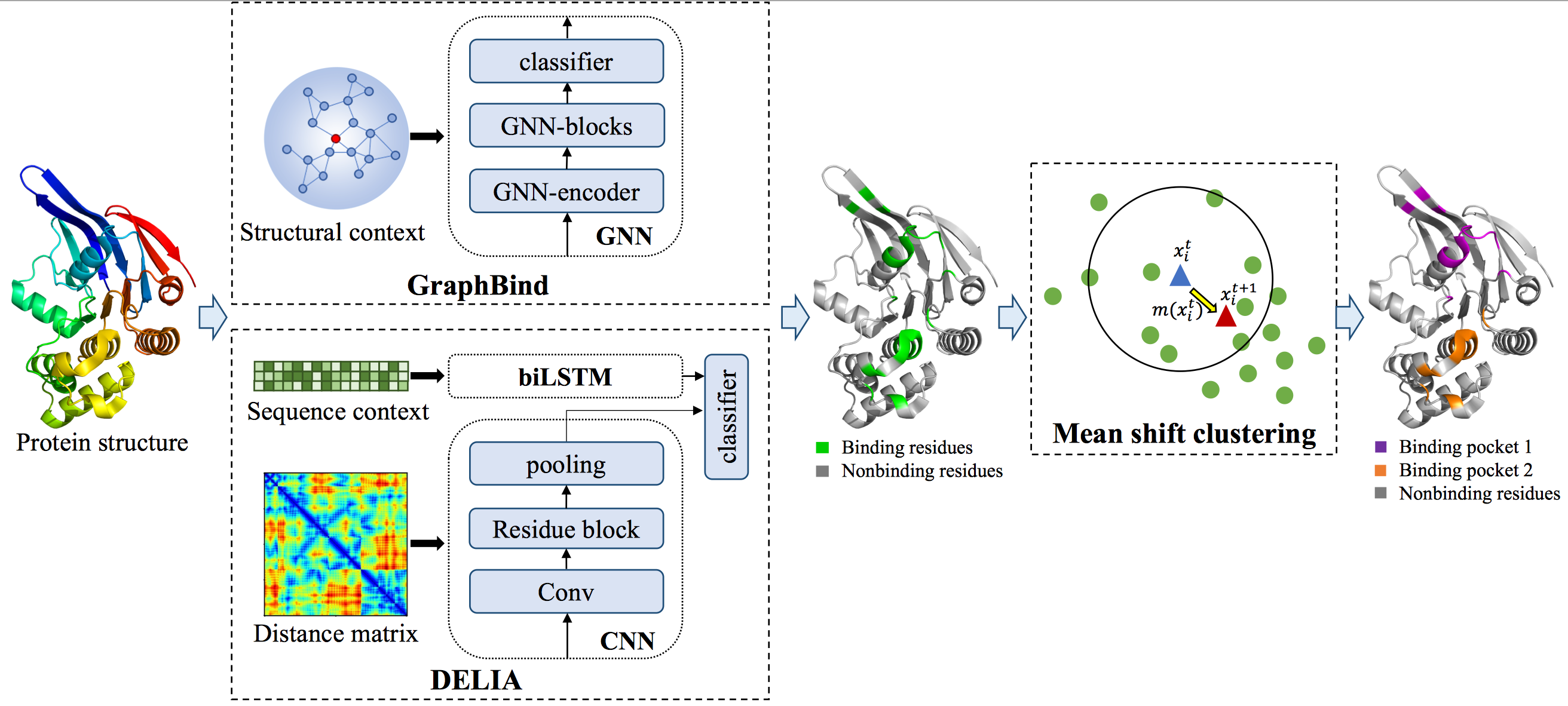
Figure 1.The flowchart of constructing a graph based on structure context in GraphBind.
GraphBind is a webserver designed for structure-based nucleic acid- and small ligand-binding residues prediction.
GraphBind consists of two modules:
1) Constructing graphs based on structure, which integrates the local neighborhood around residues to construct
graphs.
2) The hierarchical graph neural networks (HGNNs), which progressively updates the edge features, node features
and graph features, and further learns high-level features for classifying the binding residues.
Besides, we transfer the proposed HGNNs to GraphBind-G, a ligand generic model that is trained by proteins bound to hundreds of ligand types.
DELIA is a web server designed for structure-based protein-ligand binding residues prediction. Multiple deep neural networks are combined to generate the binding probability for each residue in a single protein chain. Distance matrices extracted from 3D coordinates are integrated with sequence-profiling as the input features of DELIA. DELIA has been evaluated on several benchmark datasets and the results show that it can outperform its old version and state-of-the-art methods.
Furthermore, based on the observation that the residues located in protein-ligand binding interfaces tend to form spatial clusters, we apply the mean shift clustering to further identify which of the predicted binding residues may potentially form the binding pocket(s).
2. Inputs
BindWeb only supports processing one sequence at a time. Please input the protein chain structure
(in PDB format) and
1-character chain ID (chain ID is case sensitive, if your query chain doesn't contain chain ID,
just leave the chain ID box blank). There are two functions to choose from.
You can choose the ligand-specific GraphBind and DELIA and the ligand types to predict ligand binding residues for the desired ligands.
Or you can use the ligand-general GraphBind-G to predict the binding residues for a general ligands.
Finally, you can input your Email address for receiving results.
BindWeb’s runtime is affected by its two base predictors. For ligand-general mode and ligand-specific mode for nucleic acids, the runtime of BindWeb is determined by GraphBind. For the other five ligands, the runtime is determined by DELIA.
Besides, the runtime is related to the length of query protein chain and the condition of our server.
For a protein with 100 residues, GraphBind and DELIA take approximately 10 minutes and 1 hour, respectively.
Therefore, We do not recommend you to submit a very long protein chain.
3. Outputs
We will send the results to your email when the job is finished.
Results will be shown in the result page (example) when the job is finished.
Binding residues are displayed in "Residue + Residue sequence number in PDB file" format.
For Ca2+, Mn2+, Mg2+, ATP and HEME, high confidence binding residues (the mean of predicted binding scores of GraphBind and DELIA) are highlighted in red color; medium confidence binding residues (predicted by either GraphBind or DELIA) are highlighted in blue color. For DNA, RNA and general ligands, binding residues are highlighted in red color.
The 3D structure of query protein and binding residues are also shown in the JSmol.
In addition, results can be downloaded by clicking "Download results".
This results can be saved for two weeks.
1Ying Xia, Chun-Qiu Xia, Xiaoyong Pan, Hong-Bin Shen, GraphBind: protein structural context embedded rules learned by hierarchical graph neural networks for recognizing nucleic-acid-binding residues. Nucleic Acids Research, 2021, 49(9): e51-e51.
2Chun-Qiu Xia, Xiaoyong Pan, Hong-Bin Shen, Protein-ligand binding residue prediction enhancement through hybrid deep heterogeneous learning of sequence and structure data, Bioinformatics, 2020, 36: 3018-3027.