1. Introduction
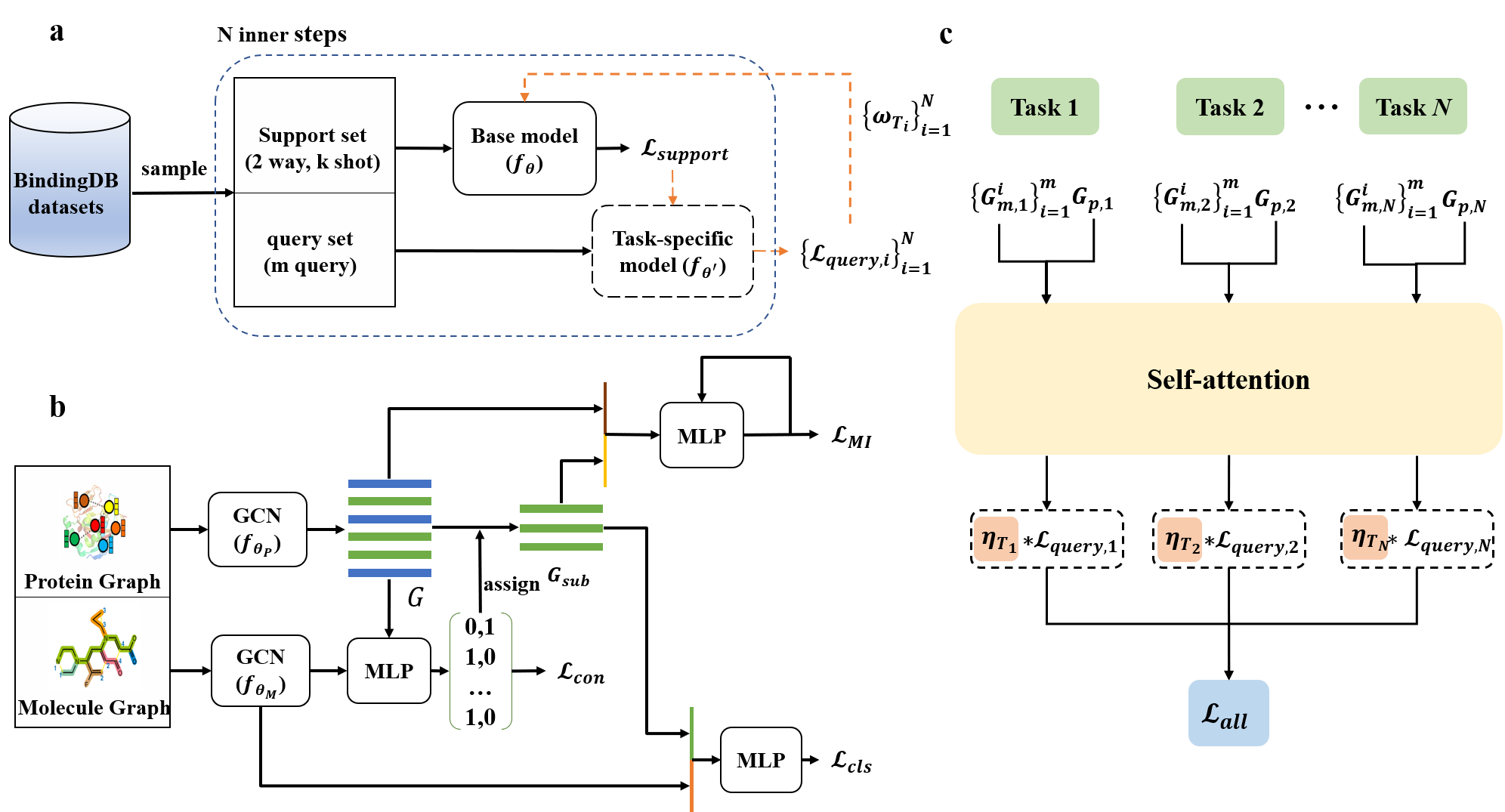
Existing drug-target interaction (DTI) prediction methods generally fail to generalize well to novel (unseen) proteins and drugs. In this study, we propose a protein-specific meta-learning framework ZeroBind with subgraph matching for predicting protein-drug interactions from their structures. During the meta-training process, ZeroBind formulates training a model per protein, which is also considered a learning task, and each task uses graph neural networks (GNNs) to learn the protein graph embedding and the molecular graph embedding. Inspired by the fact that molecules bind to a binding pocket in proteins instead of the whole protein, ZeroBind introduces an unsupervised subgraph information bottleneck (SIB) module to recognize the maximally informative and compressive subgraphs in protein graphs as potential binding pockets. In addition, ZeroBind trains the models of individual proteins as multiple tasks, whose importance is automatically learned with a task adaptive self-attention module to make final predictions. The results show that ZeroBind achieves superior performance on DTI prediction over existing methods, especially for those novel proteins and drugs, and performs well after fine-tuning for those proteins or drugs with a few known binding partners. The framework of ZeroBind is shown in Figure 1.
2. Input
First, for binding predictions of proteins and drugs, please input the protein 3D structure (in PDB format)
and molecule smiles.
Automatically, ZeroBind will search the well trained protein-specific model against the protein you input. If unfortunately our data set does not contain the protein you input, ZeroBind will turn to use the meta-model for the binding prediction task.
3. Output
We will send the results to your email when the job is finished.
Results will be shown in the result page (example) when the job is finished. In addition, results can be downloaded by clicking "Download results".